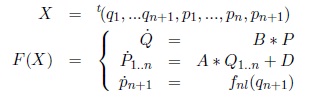INTERaction GAz-Grain
Auteurs:
Manuel Herbert , David Dalvariée, Olivier Rauquef, William Iz, Julien Trigger
Date de création :
11/10/2011
Date de mise à jour :
07/01/2012
Table des matières
Champ
Milieu interstellaire
Introduction
Le milieu interstellaire est composé d'un gaz constitué d'environ 90% d'hydrogène et 10% d'hélium, et l'intégralité de ce gaz constitue environ 99% de la matière interstellaire. Le pourcent restant correspond à la poussière interstellaire, c'est-à-dire à des "grains" composés, entre autres, de carbone et de silicium.
Le gaz du milieu interagit avec ces grains. Cette appliquette s'intéresse particulièrement aux cas des collisions entre un atome d'hydrogène du gaz et un des atomes de carbone d'un grain ou d'une chaîne carbonée.
-
Objectifs
-
L'élaboration de cette appliquette a suivi un certain nombre d'étapes :
- Création d'un système où un atome d'hydrogène est lancé, à une vitesse choisie par l'utilisateur, sur un seul atome de carbone. Il fut nécessaire de résoudre les équations différentielles correspondant au système hamiltonien associé aux deux particules, à l'aide d'un intégrateur numérique — à pas fixe dans un premier temps, puis à pas variable.
- Généralisation des équations différentielles pour la collision d'un atome d'hydrogène avec une chaîne d'un nombre quelconque d'atomes de carbone. La chaîne devrait pouvoir être choisie libre ou fixe.
- Affichage de la trajectoire des atomes en fonction du temps, de l'erreur sur l'énergie totale, et d'une animation montrant l'interaction.
Explications
Modèles
La structure carbonée
Les poussières "cibles" de l'atome d'hydrogène seront choisies comme des structures carbonées régulières. Elles sont a priori tridimensionnelles mais cette appliquette ne présentera que le cas 1D, à savoir une chaîne de n carbone fixée ou non à un bâti.
On peut s'interroger sur la pertinence d'une telle modélisation qui pourrait être qualifiée d'un peu
trop simpliste. Mais cela n'est pas complètement le cas. Bien évidemment, les équations et la programmation sont plus commodes à 1D mais ces chaînes qui semblent utopiques (la nature n'aime pas trop en général ce qui n'est pas 3D, la notion de "couche" par exemple n'est qu'une vue de l'esprit, simple modèle limite des physiciens qui conduit souvent d'ailleurs à des
discontinuités) ne le sont pas. Il existe dans la nature de véritables chaînes carbonées : les Cyanopolyynes [pour plus de renseignements voir
http://iopscience.iop.org/0004-637X/505/1/278 (en anglais) qui discute de l'existence de telles molécules dans le milieu interstellaire.] D'autres points, liés à la modélisation 1D, sont cependant difficiles à justifier physiquement : le fait que l'atome d'hydrogène vienne percuter la chaîne exactement dans l'axe par exemple...
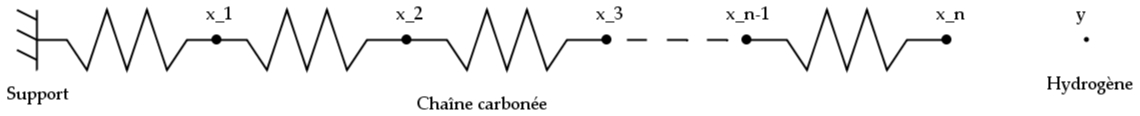
Quant à l'existence de 2 modèles chaînes : liée au bâti ou libre, elle se justifie assez bien. L'existence des Cyanopolyynes justifie déjà l'utilisation de la chaîne libre ; en ce qui concerne une chaîne liée à un bâti il suffit de considérer le bâti comme une très grosse molécule (son inertie très grande devant celle de l'atome d'hydrogène incident lui assurera de ne pas trop bouger pendant l'interaction, c'est-à-dire de jouer finalement le rôle d'une sorte de... bâti bien solide), la chaîne quant à elle sera tel un "cheveu" avec lequel viendra interagir l'atome d'hydrogène.
Les 2 figures qui suivent représentent ces 2 modèles. La position des atomes de carbone est repérée par les xn tandis que l'atome d'hydrogène incident sera associé à y.
Modèle "Chaine libre"
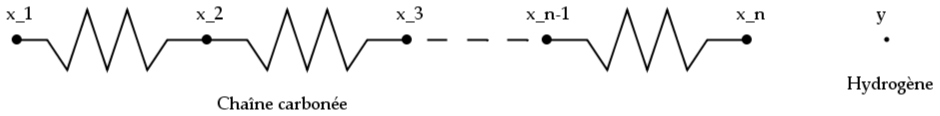
Modèle "Chaine liée à un bâti"
Les interactions entre les atomes de carbone (de masses identiques
M) sont modélisées par de simples ressorts de raideur
k. La distance inter-atomes sera notée d
CC. Le potentiel d'interaction de la chaîne de ressorts aura donc pour forme
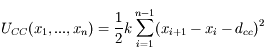
pour la chaîne libre -- à laquelle s'ajoutera
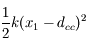
si l'on considère le modèle avec bâti.
Quant aux valeurs numériques des paramètres du système on a :
-
M, la masse du carbone qui vaut environ 12 masses atomiques.
-
k, raideur du ressort qui est calculée à partir de la formule suivante :
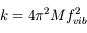 où fvib vaut environ 1012Hz qui est la fréquence typique de vibration des liaisons inter-carbones.
où fvib vaut environ 1012Hz qui est la fréquence typique de vibration des liaisons inter-carbones.
-
dCC, distance au repos entre les atomes qui est de l'ordre de 150 pm.
Les interactions Hydrogène-Carbone
On doit désormais s'intéresser à l'interaction que va subir l'hydrogène à l'approche de l'atome de carbone. On simplifie premièrement le modèle en supposant que l'hydrogène n'interagira qu'avec le dernier carbone de la chaîne (xn).
Le potentiel qui assumera l'interaction sera ensuite celui de
Lennard-Jones. Attractif aux longues distances (interactions classiques de type Coulomb), il est répulsif à très petite distance (résultat provenant de la physique quantique traduisant le principe d'exclusion de Pauli). Il existe donc une distance où ce potentiel est minimal
r0 qui correspond à la distance d'équilibre entre un atome de carbone et un d'hydrogène. Un second paramètre
E règle la profondeur de ce "puits" d'attraction.
![U_{CH}(x_n,y)=2E \left[\frac{1}{2}\left(\frac{r_0}{y-x_n}\right)^{12}-\left(\frac{r_0}{y-x_n}\right)^6\right]](eq_tex_intergag_doc/equation4.png)
Potentiel de Lennard-Jones pour r0 = 1 nm et E = 0,07 eV
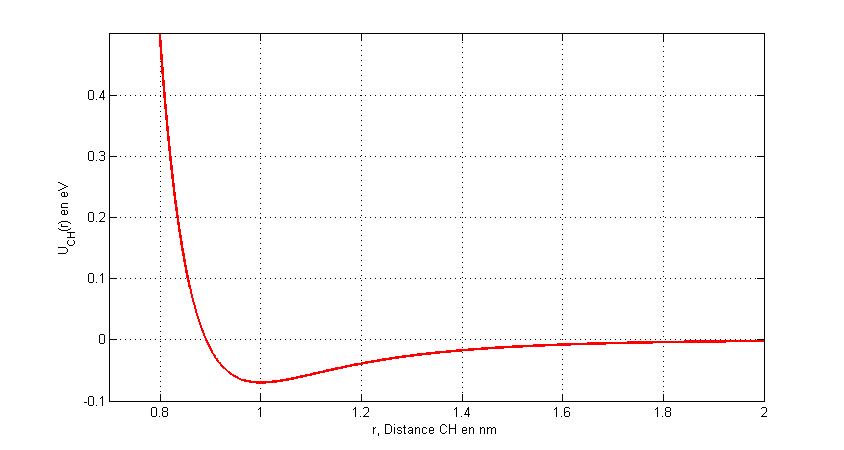
Pour les valeurs numériques typiques on a :
-
E, profondeur du puits d'attraction du potentiel de Lennard-Jones qui vaut aux alentours de 0.07 eV.
-
r0, distance d'équilibre pour le potentiel de Lennard-Jones, qui est à peine plus faible que dCC c'est à dire 0.1 nm.
Les équations du mouvement
Les potentiels définis, il est désormais possible d'écrire les équations de la dynamique. On utilisera les notations et équations de la mécanique analytique. Voici la trame générale des calculs qui permettront d'aboutir à la formulation et à la résolution du problème physique qui nous intéresse.
- Dédimensionnement du système
- Écriture du lagrangien dédimensionné
- Passage aux variables hamiltoniennes
- Équations d'Hamilton
- Résolution des équations d'Hamilton
- Retour dans l'espace réel et redimensionnement.
Energies cinétiques, potentielles et dédimensionnement
L'énergie cinétique du système correspond à la somme des énergies cinétiques de chacun des atomes :
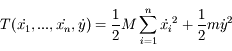
où
m est la masse de l'atome d'hydrogène (1 masse atomique).
Le potentiel total est la somme des potentiels élastiques de la chaîne de carbone et du potentiel de Lennard-Jones. Noter que l'on se placera pour la suite des calculs dans le cas du modèle "Chaîne libre". (Le calcul dans le cas "Chaîne avec bâti" ne diffère que très peu : voir plus haut la forme du potentiel lorsque le ressort "bâti-1
er atome" est pris en compte.)
![U(x_1,...,x_n,y)= \frac{1}{2}k\sum_{i=1}^{n-1} (x_{i+1}-x_i-d_{cc})^2+2E \left[\frac{1}{2}\left(\frac{r_0}{y-x_n}\right)^{12}-\left(\frac{r_0}{y-x_n}\right)^6\right]](eq_tex_intergag_doc/equation6.png)
Ces énergies cinétiques et potentielles sont dimensionnées. Un léger changement de variables est souvent pratique pour ramener ces quantités à des ordres de grandeur plus facilement maniables. On utilise pour cela les données du problème comme "grandeurs typiques". Les changements concernent ici le temps et les longueurs.
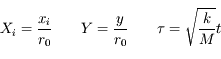
Ces changements disent simplement que la règle unité pour mesurer les distances fera une longueur de r0. Pour le temps, la pulsation de référence coïncidera précisément avec celle typique d'oscillation de la chaîne de carbone. Pour résumer on va manipuler des quantités sans dimension entre 10-1 et 10.
Lagrangien
On récupère alors un Lagrangien (L=T-U) de la forme :
![L \propto \frac{1}{2} \left[ \sum_{i=1}^n \dot{X}_i^2 + \mu \dot{Y}^2 \right] - V_0\left[\frac{1}{2}\left(\frac{1}{X_n-Y}\right)^{12}-\left(\frac{1}{X_n-Y}\right)^6\right]-\frac{1}{2} \sum_{i=1}^{n-1} (X_{i+1}-X_i-D_{cc})^2](eq_tex_intergag_doc/equation8.png)
avec
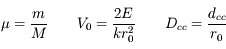
Il est ensuite nécessaire de faire apparaître les variables physiques les plus pertinentes. Par exemple dans le potentiel de Lennard Jones, deux variables sont visibles :
Xn et
Y alors que c'est toujours la quantité
Y - Xn dont dépend véritablement le potentiel. Chose tout à fait logique puisque ce potentiel est fonction de la distance entre les atomes C et H.
Globalement on va effectuer :
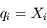
pour
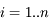
et
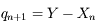
. Les quantités dérivées seront transformées de la même manière.
Hamiltonien
Le précédent changement de variable n'est pas le dernier. Les variables hamiltoniennes ne sont en effet pas
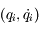
mais
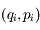
qu'on appelle
positions, impulsions. On calcule les impulsions
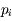
par la formule
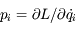
qui traduit le fait que l'impulsion est la variable conjuguée de la position.
On a alors un hamiltonien (sorte d'énergie mécanique dans les cas les plus simples comme ici)
H=T+U de la forme :
![H \propto \frac{1}{2} \sum_{i=1}^{n-1} p_i^2 + \frac{1}{2} (p_n-p_{n+1})^2+\frac{1}{2\mu}p_{n+1}^2 + V_0\left[\frac{1}{2q_{n+1}^{12}}-\frac{1}{q_{n+1}^6}\right]+\frac{1}{2} \sum_{i=1}^{n-1} (q_{i+1}-q_i-D_{cc})^2](eq_tex_intergag_doc/equation17.png)
Équations d'Hamilton
On peut finalement écrire l'équivalent du
principe fondamental de la dynamique dans l'espace d'Hamilton, résumé par les célèbres équations d'Hamilton :
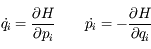
Ce qui donne, lorsqu'on les écrit pour tout i, les équations matricielles suivantes (attention elles ne sont valables que pour le modèle de la chaîne libre) :
Résolution des équations d'Hamilton
Il ne reste donc qu'à résoudre ces équations. Elles peuvent sembler un peu barbares mais en réalité elles le sont bien peu car elles sont linéaires pour la plupart des inconnues, la notation matricielle d'ailleurs le prouve et si l'on omet l'équation qui traduit l'interaction entre l'hydrogène et le carbone :
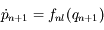
(avec
nl pour Non Linéaire) le système peut même être résolu analytiquement. Malheureusement c'est précisément cette interaction qui nous intéresse, impossible donc de l'oublier.
Le module qui va résoudre ces équations est un
intégrateur numérique (son fonctionnement sera décrit dans la section algorithmique). Celui-ci étant censé fonctionner pour n'importe quelle équation différentielle, il faut encore réduire la forme des équations du mouvement à une forme ultimement épurée à savoir :
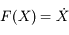
.
En fait tout est histoire de notations, il suffit en effet de définir F et X de la sorte :
Conditions initiales
Il manque cependant une chose essentielle pour résoudre les précédentes équations différentielles. Ce sont les conditions initiales ! Mon prof de maths lors du cours dédié au théorème de Cauchy-Lipschitz, disait que pour jouer à la belote, il fallait deux choses : d'abord en connaître les règles et aussi, "et surtout" insistait-il, que chacun des 4 joueurs ait sa huitaine de cartes en main. Les règles, ce sont les équations de la physique et plus généralement les équations différentielles. La distribution des cartes, quant à elle, assure la définition d'un état initial. Ces deux choses, l'une dans l'autre, permettent ensuite à la partie de se dérouler ou autrement dit au système physique d'évoluer. (Les rigoristes les plus acharnés douteront peut-être du parallèle puisque qu'à un jeu précis donné, plusieurs stratégies peuvent s'imposer ce qui rompt ainsi l'unicité de la solution qu'est censé offrir Cauchy-Lipschitz. Mais passons, c'était sur le rôle indispensable des conditions initiales qu'il fallait s'attarder.)
Il faut donc connaître ici (pour renseigner l'intégrateur numérique) position et vitesse de chacun des atomes du système à l'instant t=0.
Remarque :
Le but premier de notre appliquette était de simuler la projection d'une particule d'hydrogène de vitesse modulable sur une chaîne carbonée qui n'était pas au repos. Comprendre ici que le milieu interstellaire n'est pas complètement calme. Un rayonnement électromagnétique y est toujours présent. Et celui-ci doit en principe exciter la chaîne dans un certain mode de vibration. La chaîne peut également rayonner lorsqu'elle est trop excitée. Il en résulte qu'à l'équilibre thermique (égalité entre l'absorption et l'émission de la chaîne), il est possible de définir une température typique pour la chaîne, liée directement à l'énergie de celle-ci et donc à son mode d'oscillation. Ce paramètre de température n'a malheureusement pas encore été programmé. C'est pourquoi on a supposé dans cette appliquette que la chaîne de ressorts était au repos initialement.
-
Chaîne libre initialement au repos : On placera bien évidemment chaque atome de carbone distant de dcc de ses voisins. Il y a donc un seul arbitraire : la position du centre de gravité de la chaîne. Et celle-ci sera fixée à l'origine à l'instant t=0. Quant aux vitesses, elles sont toutes choisies nulles.
-
Chaîne liée au bâti initialement au repos : Cette fois-ci, c'est le bâti qui servira d'origine. Les atomes seront ensuite placés les uns après les autres en respectant la distance d'équilibre dcc. Les vitesses sont encore une fois choisies toutes nulles.
-
Particule d'hydrogène : L'appliquette fixe initialement la particule d'hydrogène à 5r0 du dernier atome de carbone. A cette distance le potentiel de Lennard Jones ne se fait pas encore trop sentir, de cette manière la particule incidente a un trajectoire quasi rectiligne avant de "plonger" et d'interagir avec le carbone. Pour résumer, cette distance initiale permet de voir la transition "trajectoire rectiligne uniforme - trajectoire accélérée". Quant à la vitesse v0, l'utilisateur pourra la fixer selon son bon vouloir. Elles devront cependant être négatives pour pouvoir se rapprocher du dernier atome de carbone (sinon pas d'interaction !). Précisons de même que leur norme typique va de 500 à 1500 ms-1.
Voici le vecteur (position, vitesse) initial pour la chaîne libre :
![\left\{ (x_i(0),\dot{x_i}(0))=\left(\frac{d_{cc}}{2}[2i-(n+1)],0\right) \right\}_{i=1..n}](eq_tex_intergag_doc/equation21.png)
.
Et pour l'hydrogène :
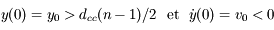
.
Il reste une petite subtilité pour fournir ces données à l'intégrateur. En effet, les équations différentielles étant dans l'espace d'Hamilton, il faut passer les conditions initiales dans ce même espace. A savoir faire suivre la cheminement
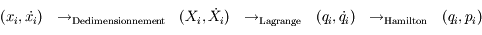
aux conditions initiales précédemment explicitées. Ce qui donne, en passant les calculs :
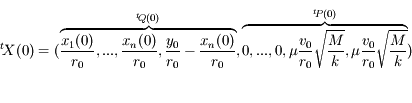
Ce qu'on fournit à l'intégrateur numérique est donc la fonction F et ce vecteur initial X(0). Il renverra le vecteur X(t).
Retour dans l'espace réel et redimensionnement
Il restera alors simplement à faire revenir ce vecteur
X(t) qui sera dans l'espace d'Hamilton, dans l'espace réel. Il n'y aura qu'à suivre exactement le chemin inverse de tout à l'heure. Connaissant donc
X c'est à dire les
qi et les
pi à chaque instant, on en déduira les positions et vitesses de chaque atome grâce aux formules :
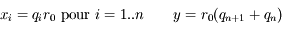
pour les positions
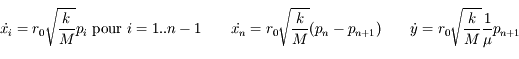
pour les vitesses.
Intégrateur numérique
L'intégrateur numérique est un outil qui sert à résoudre une équation différentielle donnée. Dans notre cas (étude de la dynamique d'un système à plusieurs corps) la mise en équations du problème aboutit à un système d'équations différentielles couplées. Cependant, si elle ne contient pas de termes en
y', on peut toujours transformer une équation différentielle du second ordre en deux équations différentielles du premier ordre via l'introduction d'une variable intermédiaire :
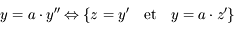
.
Il suffit donc que l'intégrateur numérique soit capable de résoudre un système d'équations différentielles du premier ordre pour pouvoir résoudre n'importe quel système d'équations différentielles linéaires.
On veut donc résoudre un système du type :
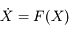
où
X est une grandeur vectorielle.
Méthode de Runge-Kutta
La méthode de Runge-Kutta peut être vue comme une amélioration de la méthode d'Euler. Dans la méthode d'Euler, on résout numériquement une équation différentielle de type
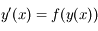
de la façon suivante : on connaît une condition initiale
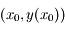
. On peut donc calculer
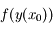
qui n'est autre que
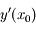
. En choisissant un
pas d'intégration h on peut donc déterminer approximativement
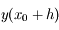
:
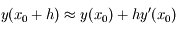
Connaissant
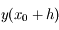
on peut ensuite évaluer
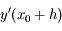
, puis calculer
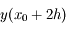
grâce à la formule précédente et ainsi de suite...
Bien qu'assez simple, cette méthode accumule une erreur assez importante rapidement car l'erreur lors du calcul de
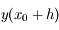
est d'ordre
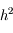
. On cherche donc à évaluer de façon plus précise
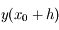
. La méthode de Runge-Kutta d'ordre 2 consiste à évaluer
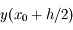
par une méthode d'Euler et à utiliser ensuite cette valeur pour calculer la dérivée
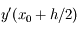
. On calcule ensuite
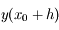
par :
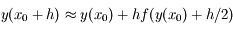
On peut prouver que l'erreur commise sur
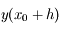
dans ce cas est d'ordre
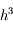
, on peut par ailleurs se convaincre du fait que cette méthode est plus efficace en traçant un graphe (avec une fonction
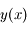
convexe par exemple). Bien que l'erreur commise sur une itération soit d'ordre
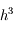
, cette méthode s'appelle
RK2 car l'erreur commise sur toute la durée d'intégration est
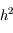
.
On voit donc l'utilité de la méthode de Runge-Kutta : on double le nombre de calculs nécessaires (puisqu'il faut évaluer deux fois la dérivée) mais on gagne un ordre sur la précision. Il est ensuite possible de faire plus d'évaluations intermédiaires de la dérivée afin d'obtenir des erreurs encore plus faibles. Par exemple la méthode
RK4, où :
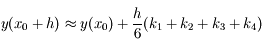
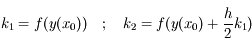
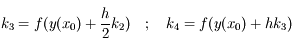
permet d'obtenir une erreur en
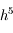
sur le pas, d'où la dénomination
RK4. C'est cette méthode qui est utilisée dans le code.
Pas variable
On peut maintenant intégrer des systèmes d'équations différentielles avec une bonne précision mais un problème persiste : le pas d'intégration
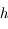
doit être fixé à l'avance. Or, pour une trajectoire
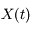
donnée, on va accumuler une erreur importante quand les dérivées n-ièmes ont une grande valeur, et au contraire on va commettre peu d'erreur quand les dérivées n-ièmes sont petites. Pour chaque itération de l'intégration on va chercher le pas
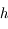
engendrant une erreur fixée à l'avance par l'utilisateur.
Le principe est donc le suivant (on considère une fonction de la variable temporelle
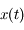
solution de l'équation différentielle
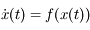
: à partir d'une position initiale
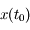
on calcule
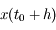
en faisant un pas de taille
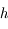
(on note
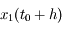
) et on calcule
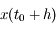
en faisant deux pas de taille
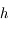
(on note
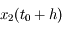
). L'erreur commise sur
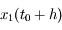
est
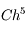
où
C est une constante et l'erreur commise sur
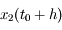
est
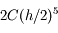
. On a donc
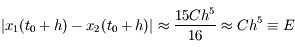
.
Si maintenant l'utilisateur à fixé une erreur relative sur la précision
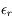
, l'erreur absolue vaut
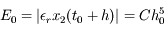
.
est donc le pas permettant d'obtenir l'erreur relative
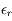
. On obtient donc au final :
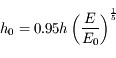
Le 0.95 permet de se laisser une marge de sécurité pour être sûr d'avoir une précision suffisante, puisque
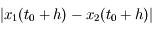
n'est pas exactement égal à
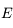
.
Une fois
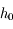
calculé on refait toute la procédure jusqu'à ce que l'erreur commise avec
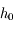
soit inférieure à l'erreur souhaitée. On peut ensuite calculer
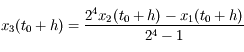
ce qui permet d'avoir une erreur sur le pas en
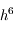
, et donc globalement un
RK5.
Méthodes JAVA
L'implémentation de cet intégrateur est contenue dans une classe JAVA "intégrateur". Cette classe contient une méthode rk4 qui calcule une itération à la méthode RK4, une méthode rk4pv qui calcule une itération avec le pas optimal pour l'erreur demandé, et une classe rk4dt qui effectue une intégration sur une période T en tronquant le pas variable de sorte à ce que l'intégration finisse exactement au temps T. Ceci est nécessaire pour pouvoir afficher les points des solutions à intervalles réguliers.
Mode d'emploi de l'applet
L'utilisateur, en vue de tester différentes situations, pourra à sa guise modifier les paramètres suivants :
- La vitesse de l'atome d'hydrogène (en m.s-1). Elle est obligatoirement négative — l'atome partant d'une position positive pour aller vers la chaîne carbonée.
- L'énergie du puits
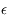 (en électon-volts). Il fixe la profondeur du puits du potentiel utilisé (celui de Lennard-Jones).
(en électon-volts). Il fixe la profondeur du puits du potentiel utilisé (celui de Lennard-Jones).
- La distance du puits de potentiel (en nm). C'est la distance à laquelle la valeur du potentiel (
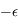 ) est la plus faible.
) est la plus faible.
- Modèle de la chaîne carbonée. La chaîne peut être choisie libre ou bien fixe.
- La durée simulée (en ps).
- Le nombre de points affichés pour effectuer le tracé.
- L'erreur maximale faite dans la classe intégration.
- Le temps d'exécution minimal (en s).
- Le choix d'algorithme. Il est nécessaire pour l'intégration et on peut le choisir à pas fixe ou à pas variable.
Après avoir déterminé les paramètres, l'utilisateur a la possibilité de tracer les trajectoires des différents atomes en fonction du temps, avec en plus une animation de la collision, ou bien avec l'erreur sur l'énergie.
Exemple d'utilisation
1 carbone fixé à un bâti
On ne peut pas faire plus simple avec ce premier exemple. Un atome d'hydrogène se dirige vers l'atome de carbone en suivant une trajectoire rectiligne uniforme. Dès lors qu'il est assez près (de l'ordre du rayon d'action du potentiel de Lennard-Jones, il interagit fortement avec lui tout en restant à distance (le potentiel infini pour une distance nulle empêche l'hydrogène de trop se rapprocher). Après un certain temps, il s'échappe en ayant transféré un peu d'énergie à la chaîne de carbone (remarquer la légère oscillation).
Interaction 1C - H
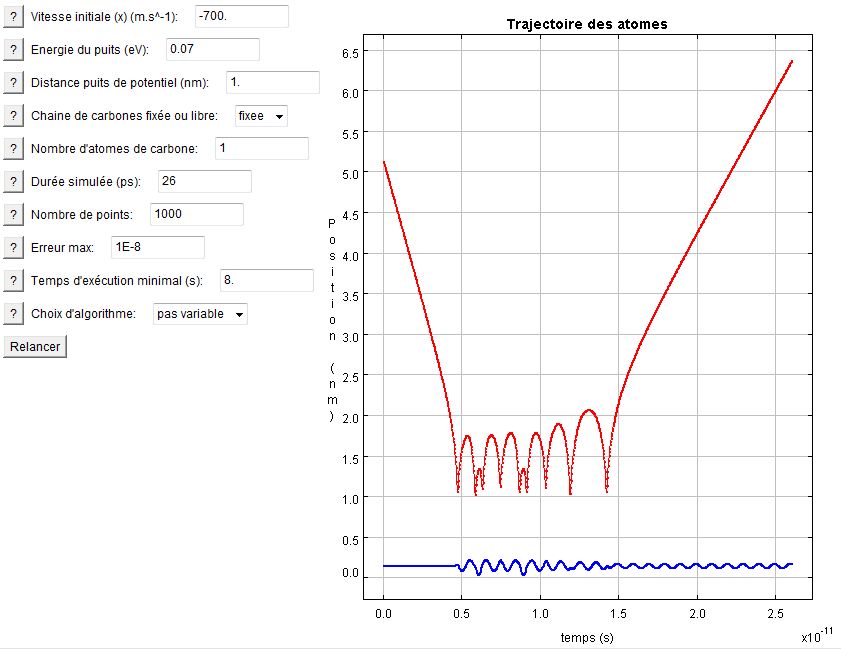
Exemple d'utilisation
Illustration du chaos
On s'aperçoit assez rapidement en jouant avec les paramètres initiaux que de faibles changements sur ceux-ci peut avoir de grandes conséquences. Le système physique auquel nous nous intéressons peut donc être qualifié de chaotique. Un infime changement de vitesse ou même parfois un léger changement sur l'erreur de l'intégrateur numérique peuvent être très influents. Ceci peut être très inquiétant si l'on ne connaît pas l'existence d'un lemme fondamental en simulation de système chaotique qui se nomme le "Lemme de l'ombre" (shadowing lemma). Car où se cache la vérité, j'entends ce qui est vraiment physique, si l'on sait que les petites erreurs numériques faites par l'ordinateur peuvent avoir d'immenses conséquences sur le résultat obtenu ? Et bien, il ne faut pas vraiment s'en soucier : ce résultat obtenu correspond bel et bien à quelque chose de physique. Cependant, il ne correspond pas aux conditions initiales rentrées. Pour résumer, il faut se dire : "Ce que j'obtiens comme résultat final correspond bien d'un point de vue physique à un jeu de données initiales mais celles-ci ne sont pas celles que j'ai choisies..."
Interaction 1C - H, autre comportement
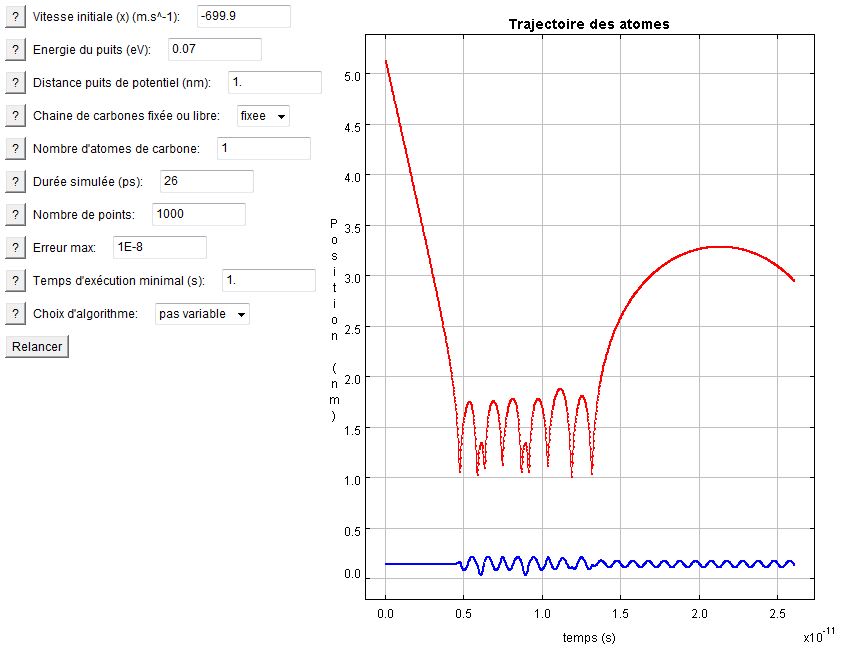
Exemple d'utilisation
2 Carbones libres
L'exemple suivant s'intéresse à l'interaction entre une chaîne libre de 2 carbones et un atome d'hydrogène. Des calculs assez simples montreraient que le comportement d'un tel système est quasiment identique au précédent : si l'on se place dans le référentiel barycentrique et que l'on ajuste la vitesse de la particule incidente, on obtient exactement le même système différentiel qu'avec 1 atome de carbone fixé à un bâti. Ce qu'il faut retenir, c'est que le comportement dépend essentiellement du nombre de ressorts. On ne s'étonne donc pas de voir le même graphe que pour le premier exemple. Cependant une chose intéressante qui n'y est pas présent est le mouvement global transféré à la chaîne par l'hydrogène incident. Il y a donc un transfert d'énergie autant dans l'oscillation que dans le mouvement du barycentre.
Interaction 2C - H
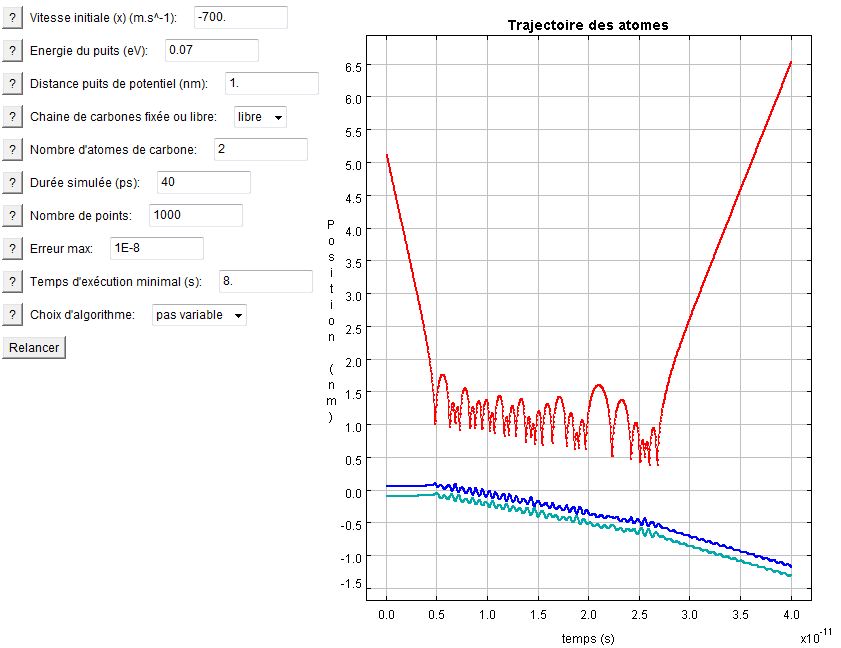
Exemple d'utilisation
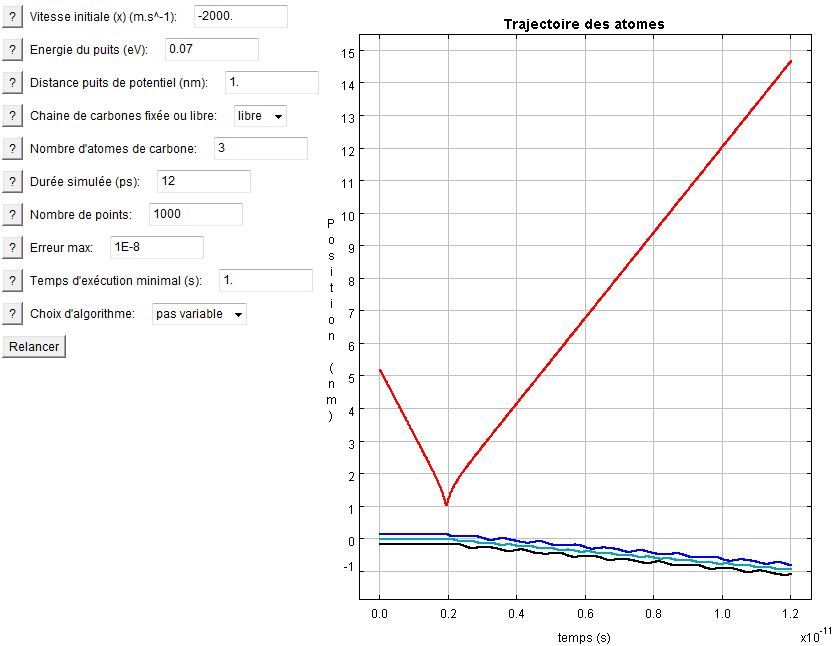
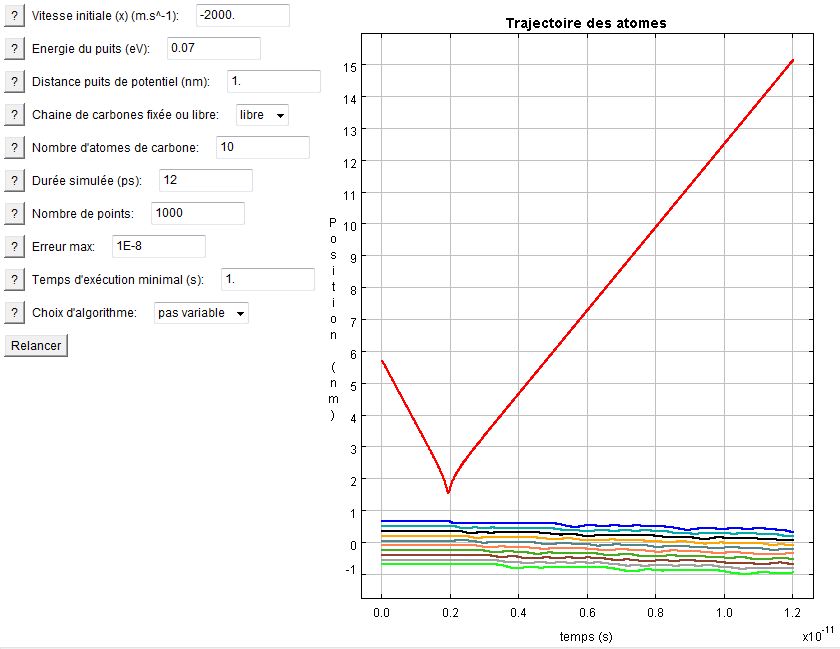
Chaînes de carbone libre
Si l'on augmente le nombre de carbone dans la chaîne, son inertie augmente. Il devient donc plus difficile pour l'atome d'hydrogène de transférer de la quantité de mouvement au barycentre. C'est ce qui est flagrant sur les deux graphes suivant. On voit clairement que la pente pour une chaîne de 3 carbones et bien plus raide que pour celle de 10 carbones. Le cas limite est évidemment une chaîne de masse infinie (pente nulle) modélisée simplement par la chaîne fixée à un bâti.
Interaction 3C - H
Interaction 10C - H
Exemple d'utilisation
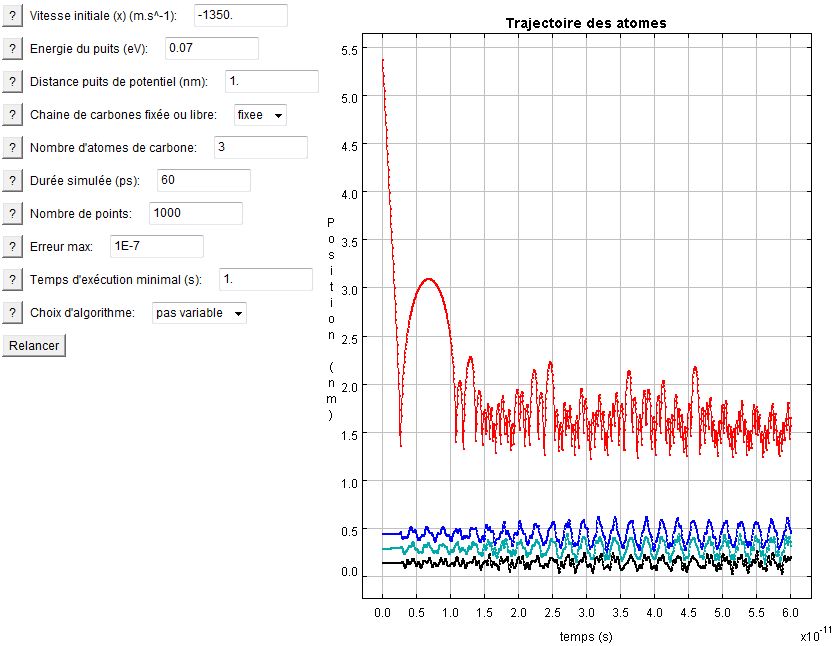
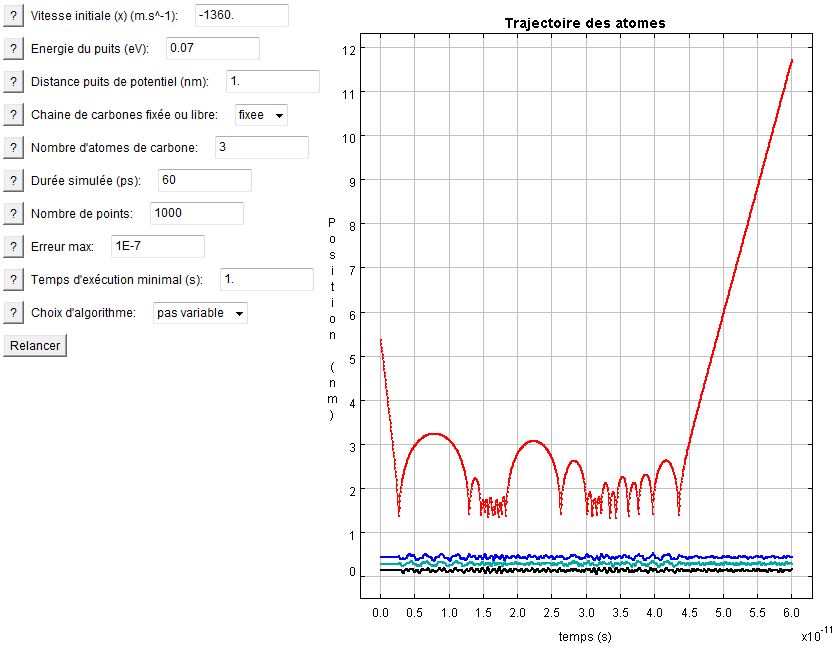
Capture ou non ?
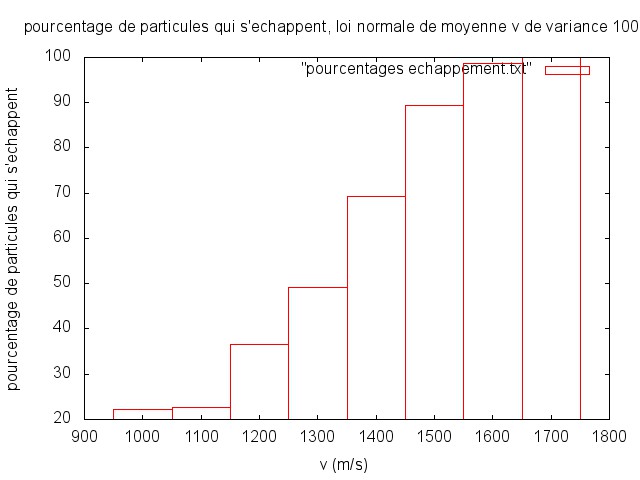
On peut se demander, étant donné que le potentiel de Lennard-Jones possède un puits de potentiel, s'il arrive que, quelquefois, l'hydrogène ne reparte pas. Et il s'avère que c'est le cas, et même souvent lorsque la chaîne est longue et que les vitesses sont faibles. A nombre de carbone fixé, l'utilisateur pourra s'amuser à déterminer la vitesse limite d'échappement. Les graphes ci-dessous illustre une capture et un échappement.
Il est parfois extrêmement difficile de savoir si l'atome va oui ou non s'échapper et le comportement chaotique accroît les difficultés de prédiction... L'atome d'hydrogène peut parfois interagir de longs moments, laissant présager qu'il est capturé, alors qu'il finit curieusement par s'échapper... Deux articles (
http://www.phchem.uni-duisburg-essen.de/photochem/preprints/Kim_001.pdf et "Hydrogen vibrational modes on graphene and relaxation of the C–H stretch
excitation from first-principles calculations" - Sakong et Kratzer [J. Chem. Phys. 133, 054505 (2010)]) discutent sérieusement de ce problème de capture et cela non pas pour une chaîne de carbone mais pour une nappe, type graphène.)
Capture v0=-1350 m.s^-1
Echappement v0=-1360 m.s^-1
Exemple d'utilisation
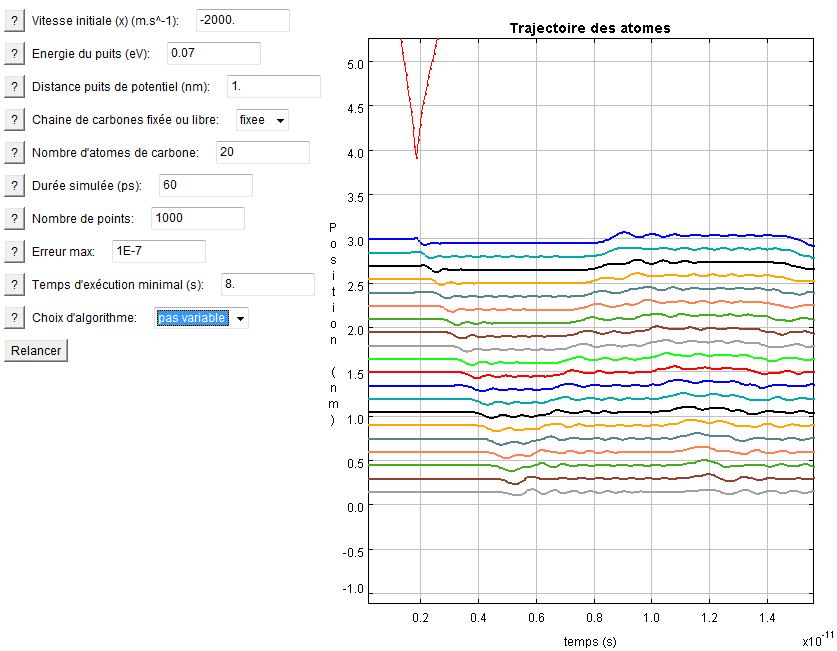
Répartition de l'énergie dans la chaîne de carbone
Le dernier exemple concerne la répartition de l'énergie dans une chaîne de carbone après un choc. On fait pour cela arriver sur une longue chaîne une particule très véloce. Celle-ci s'échappe sans interagir très longtemps, si bien que la chaîne voit cette interaction comme un choc (une impulsion diront certains, d'autres un Dirac même). Cette perturbation initiale va ensuite se propager dans la chaîne, arriver de l'autre côté, rebondir, revenir, etc. Ce qui est intéressant de remarquer c'est que, petit à petit, les oscillations finissent par se retrouver dans toute la chaîne. Ceci illustre parfaitement le fait que la chaîne est un milieu dispersif.
Dispersion d'un choc dans la chaîne
Exemple d'utilisation
Les erreurs
Il existe plusieurs moyens pour vérifier la fiabilité de l'intégrateur numérique. On peut par exemple s'assurer que le barycentre du système tout entier ne change pas de position (c'est un système isolé !). L'autre quantité qui a été choisie résulte de la
conservation de l'énergie (justifiée par le fait que l'hamiltonien ne dépende pas du temps ou que toutes les forces soient conservatives.) Il est donc assez aisé de construire une quantité qui rendra compte de cette conservation.
Tous les calculs numériques étant fait dans l'espace d'Hamilton, on utilisera l'équivalent Hamiltonien de l'énergie qui n'est rien d'autre que l'hamiltonien
H lui-même. Plutôt qu'une longue explication, voici la quantité qui permettra de s'assurer de cette conservation :
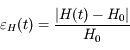
qui n'est rien d'autre que l'erreur relative entre l'hamiltonien initial et l'hamiltonien à l'instant
t.
L'utilisateur remarquera assez rapidement que cette erreur diminue lorsque la précision de l'intégrateur est accrue. (Ce qui est bien rassurant !)
Erreur sur l'énergie pour l'intégrateur "pas variable", précision souhaitée 1E-5
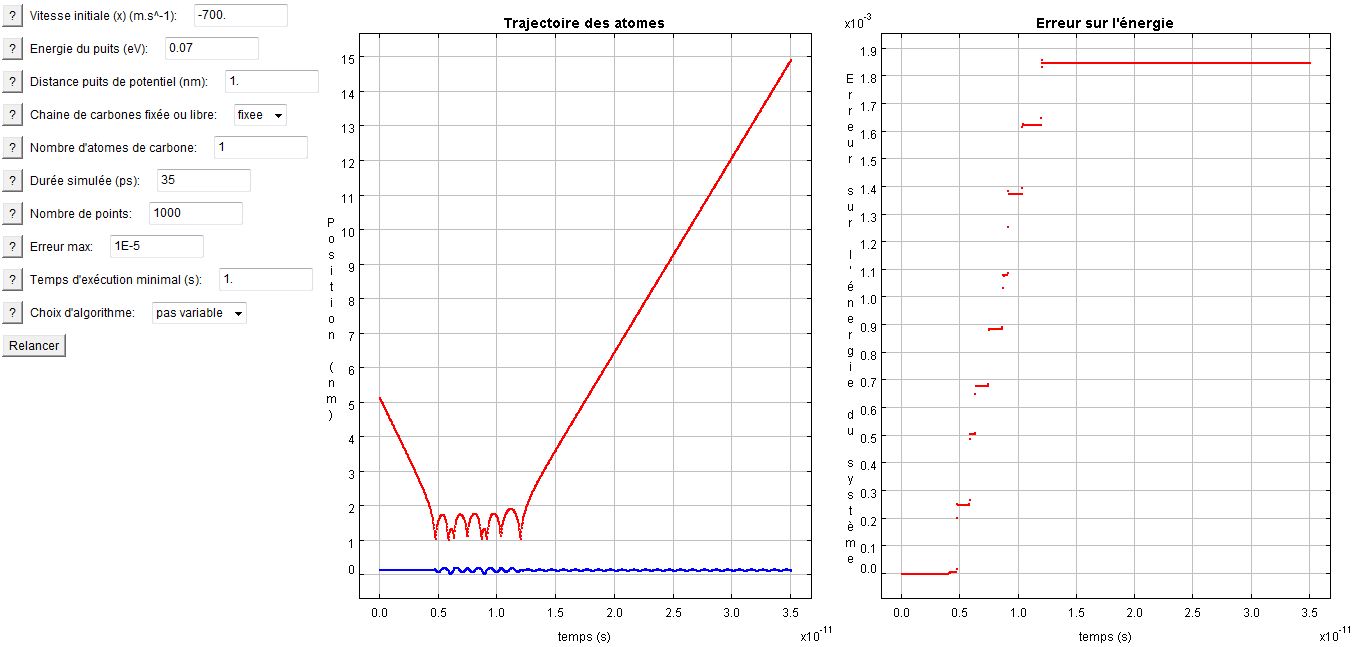
Erreur sur l'énergie pour l'intégrateur "pas variable", précision souhaitée 1E-10
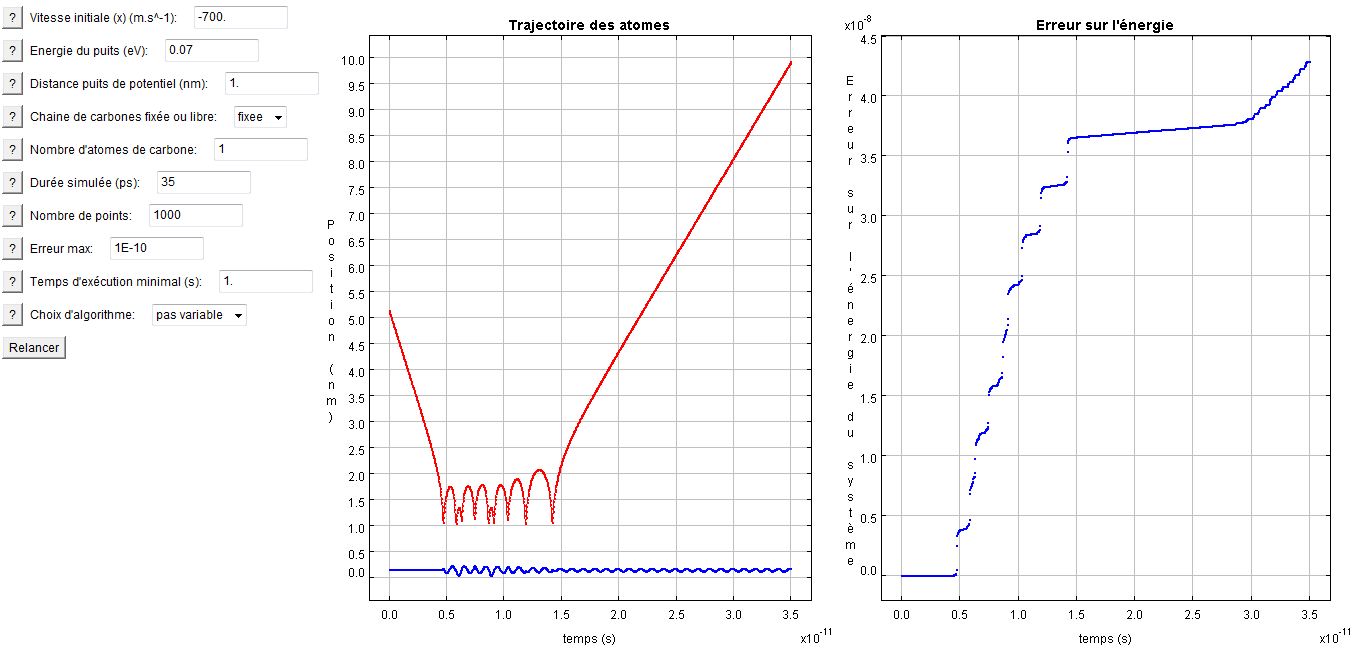
Remarque :
Sur ces deux graphes on voit bien que ce sont les non-linéarités (interactions entre hydrogène et carbone) qui créent de l'erreur. Lorsque la trajectoire de l'hydrogène est rectiligne et uniforme, l'erreur ne croît en effet quasiment pas.
Exemple d'utilisation
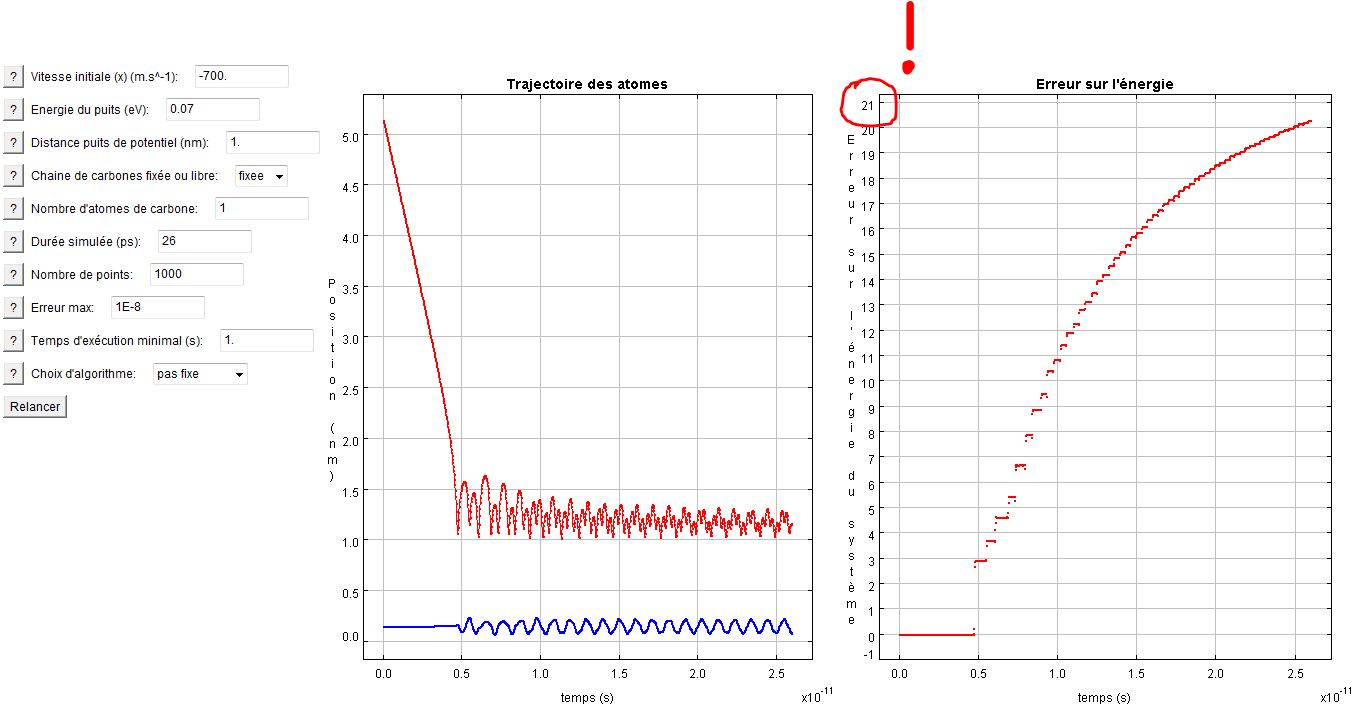
Un autre intégrateur peut être choisi par l'utilisateur. Celui-ci n'adapte pas son pas de résolution à la variation locale, autrement dit il travaille à pas fixe. L'utilisateur peut s'apercevoir que les résultats qu'il donne sont très médiocres voire complétement faux notamment lorsque l'atome d'hydrogène interagit avec le carbone, c'est-à-dire là où les vitesses changent le plus : l'hydrogène étant dans le champ du potentiel de Lennard-Jones, il subit des accélérations notoires sur des temps courts ce qui nécessite un "zoom" de l'intégrateur (ce qui est impossible car il travaille à pas fixe).
Erreur sur l'énergie avec l'intégrateur "pas fixe"
Exemple d'utilisation
Répartition des vitesses - Principe général
Dans cette partie nous allons présenter quelques résultats obtenus grâce à des essais de nature statistique. Le principe général est simple : si on veut savoir quelle est, par exemple, l'énergie moyenne laissée par une particule à la chaine carbonnée, on génère aléatoirement un grand nombre de particules ayant une distribution aléatoire de vitesses en accord avec le modèle physique, et on calcule la moyenne de l'énergie laissée à la chaine carbonnée pour chacune de ces particules.
Détection
Pour pouvoir exploiter les données résultantes des essais statistiques on a besoin de savoir à quel moment exactement l'hydrogene se libère de la chaîne carbonée (si elle s'échappe). Une technique simple consiste à fixer une distance chaîne-hydrogène assez grande (10 nm par exemple), et dire que si la distance chaîne-hydrogène dépasse 10 nm alors la particule s'est échappée. Cette méthode présente deux inconvénients : premièrement, dans certains cas l'hydrogène peut monter assez haut tout en restant lié a la chaine. Et deuxièmement, si une particule s'échappe de la chaîne mais à une vitesse faible, elle n'aura pas le temps d'atteindre les 10nm avant la fin de l'intégration et on aura dont un faux négatif.
Une meilleure technique consiste à regarder l'énergie de l'atome d'hydrogène à chaque instant. Si la somme du potentiel de Lennard-Jones et de l'énergie cinétique de l'hydrogène devient positive, c'est que l'hydrogène à une vitesse supérieure à la vitesse de libération. Seulement, ce résultat est légèrement erronné : l'hydrogène n'évolue pas dans un potentiel stationnaire et il pourrait donc se faire "rattraper" par un atome de carbone qui l'attirerait à nouveau dans son potentiel même si à un moment la vitesse de libération semblait suffisante. Pour palier ce problème on dégage une période moyenne pour le dernier atome de carbone et on effectue le test d'énergie durant une demie période. En pratique cela marche car on fait les tests d'énergie pour les positions extrémales de l'hydrogène.
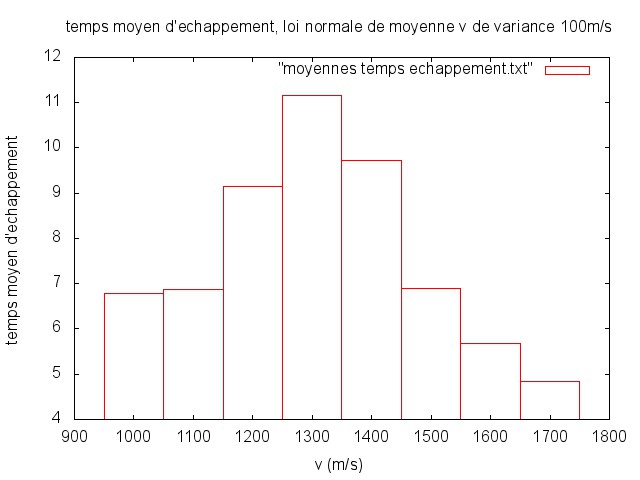
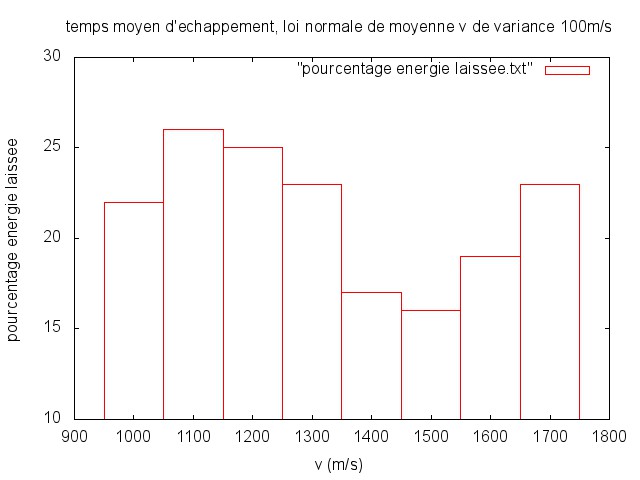
Pourcentages d'échappement, distance moyenne d'échappement et énergie laissée à la chaine
Voici quelques résultats obtenus :
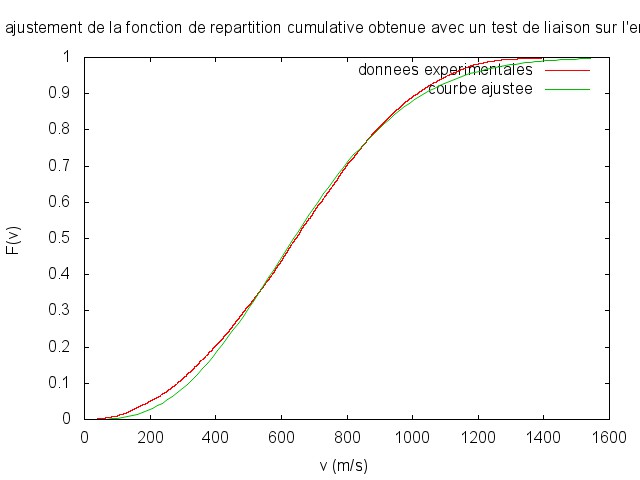
Test de la loi de probabilité des particules échappées
Dans le milieu interstellaire, il existe un loi de probabilité pour le module de la vitesse d'une particule donnée. Une particule incidente sur la chaîne de carbone aura donc une vitesse qui suit cette loi et on peut se demander si on retrouve la même loi, avec espérances et variances différentes, pour la vitesse des particules s'échappant du système. Pour ce faire, on trace la densité cumulée pour les vitesses de sortie, on ajuste un modèle correspondant à la loi que l'on veut tester et on applique le test de Kolmogorov : si le maximum
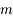
de la distance entre les courbes est tel que (
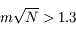
, N est le nombre d'essais), alors on peut rejeter l'hypothèse comme quoi les vitesses en sortie suivent la même loi qu'en entrée avec une confiance de 95%.
En pratique c'est bien ce que l'on obtient :
Liste des paramètres d'entrée de l'applet
-
titre : Vitesse initiale (x)
label : v0
unités : m.s^-1
Vitesse initiale de la particule suivant les x
-
titre : Energie du puits
label : eps
unités : eV
Coefficient du potentiel d'interaction fixant la profondeur du puits de potentiel
-
titre : Distance puits de potentiel
label : r0
unités : nm
Distance caractéristique du puits de potentiel
-
titre : Chaine de carbones fixée ou libre
label : typechaine
Choix du modèle pour la chaine de carbone : elle peut être libre, ou fixée à un bâti pour modéliser l'attache à une molécule plus grosse.
Valeurs possibles :
fixee
libre
-
titre : Nombre d'atomes de carbone
label : nc
Nombre d'atomes de carbone
-
titre : Durée simulée
label : tmax
unités : ps
Durée simulée
-
titre : Nombre de points
label : n_iter
Nombre de points
-
titre : Erreur max
label : errmax
Erreur d'intégration instantanée max
-
titre : Temps d'exécution minimal
label : temps_total_min
unités : s
Temps d'exécution minimal à appliquer, pour que l'affichage soit perceptible
-
titre : Choix d'algorithme
label : choixalgo
Choix de l'algorithme (pas fixe ou variable)
Valeurs possibles :
pas variable
pas fixe
Liste des paramètres de sortie de l'applet
-
titre : temps (s)
label : t
unités : s
-
titre : Position (nm)
label : xt
unités : nm
-
titre : Erreur sur l'énergie du système
label : err_energie
unités : sans unité
-
label : yt
-
titre : Position du n-ième carbone (nm)
label : xc1
unités : nm
-
label : yc1